Wow, it's been busy. Got a new ICP mass spec with some new features not seen before on any previous ICP mass spectrometers. Now, for the first time ever, copper can be analyzed in a matrix of salt water without any matrix removal, all while keeping the detection limit below the EPA action limit. Also, no internal standard suppression more than 60%. Since we don't write papers for journal submission, I'm announcing this to the chemistry community here.
Important factors in the analysis include a specific nebuliser optimization, cones that are water cooled, but are allowed to be at a higher temperature than previous models, and a really long rinse time.
Any chemist out there that are analyzing copper in seawater on ICPMS, inquire here for smooth analyses.
Thursday, February 22, 2007
Thursday, January 04, 2007
Check out Notre Dame's ICPMS lab
This place looks pretty sweet, except for the exhaust tubes hanging all over the place. If they analyze samples for free, can I sub-contract?
 The Element 2 High-Resolution ICPMS is from the good ole boys at Thermo, who have now merged with Fisher Scientific to form a massive company that can sell you anything from post it notes to 1-Aryl-2-Pyrrolidinones. With more resolution like this, you might be able to tell the difference between 32S-33S and 65Cu. Did you know that there is a lot of sulfur in seawater, but not a lot of copper? Makes it hard to analyze for copper. Oh ya, mass 63 doesn't work either because of ArNa :)
The Element 2 High-Resolution ICPMS is from the good ole boys at Thermo, who have now merged with Fisher Scientific to form a massive company that can sell you anything from post it notes to 1-Aryl-2-Pyrrolidinones. With more resolution like this, you might be able to tell the difference between 32S-33S and 65Cu. Did you know that there is a lot of sulfur in seawater, but not a lot of copper? Makes it hard to analyze for copper. Oh ya, mass 63 doesn't work either because of ArNa :)
 The Element 2 High-Resolution ICPMS is from the good ole boys at Thermo, who have now merged with Fisher Scientific to form a massive company that can sell you anything from post it notes to 1-Aryl-2-Pyrrolidinones. With more resolution like this, you might be able to tell the difference between 32S-33S and 65Cu. Did you know that there is a lot of sulfur in seawater, but not a lot of copper? Makes it hard to analyze for copper. Oh ya, mass 63 doesn't work either because of ArNa :)
The Element 2 High-Resolution ICPMS is from the good ole boys at Thermo, who have now merged with Fisher Scientific to form a massive company that can sell you anything from post it notes to 1-Aryl-2-Pyrrolidinones. With more resolution like this, you might be able to tell the difference between 32S-33S and 65Cu. Did you know that there is a lot of sulfur in seawater, but not a lot of copper? Makes it hard to analyze for copper. Oh ya, mass 63 doesn't work either because of ArNa :)
Sunday, November 26, 2006
Search for ICPMS on digg.com yields no results
I searched for "mass spectrometry" on the user powered news site, digg.com, and got no results. First, I checked under the science topic. Oh, wait there are some stories that come up when the whole site is searched.
Fairly interesting stuff, not newsworthy according to the digg community, but I don't think there are many scientist on there.
At Georgia Tech, this is what they call a nanoscale probe, or the Scanning Mass Spectrometry probe (SMS). It seems like what they are trying to do is take some mass spectrums of protiens, metabolites, and peptides without separating them from the cell/tissue. The associate professor responsible for doing this work is Andrei G. Fedorov. His research is pretty hardcore, talking about crazy ideas for ion sources, see project 7.
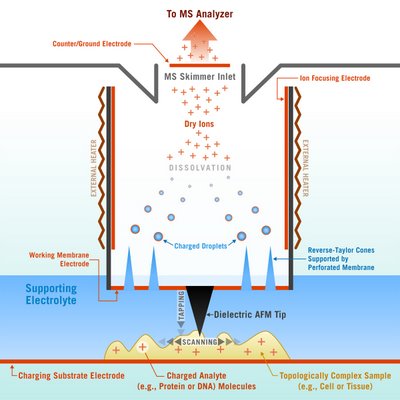
I think the way it works is the substrate gets a positive charge, the scanning tip has a negative charge. The scanning tip pulls molecules by charge, but also uses something called Taylor electrohydrodynamic focusing of jets to produce charged ions.
Fairly interesting stuff, not newsworthy according to the digg community, but I don't think there are many scientist on there.
At Georgia Tech, this is what they call a nanoscale probe, or the Scanning Mass Spectrometry probe (SMS). It seems like what they are trying to do is take some mass spectrums of protiens, metabolites, and peptides without separating them from the cell/tissue. The associate professor responsible for doing this work is Andrei G. Fedorov. His research is pretty hardcore, talking about crazy ideas for ion sources, see project 7.
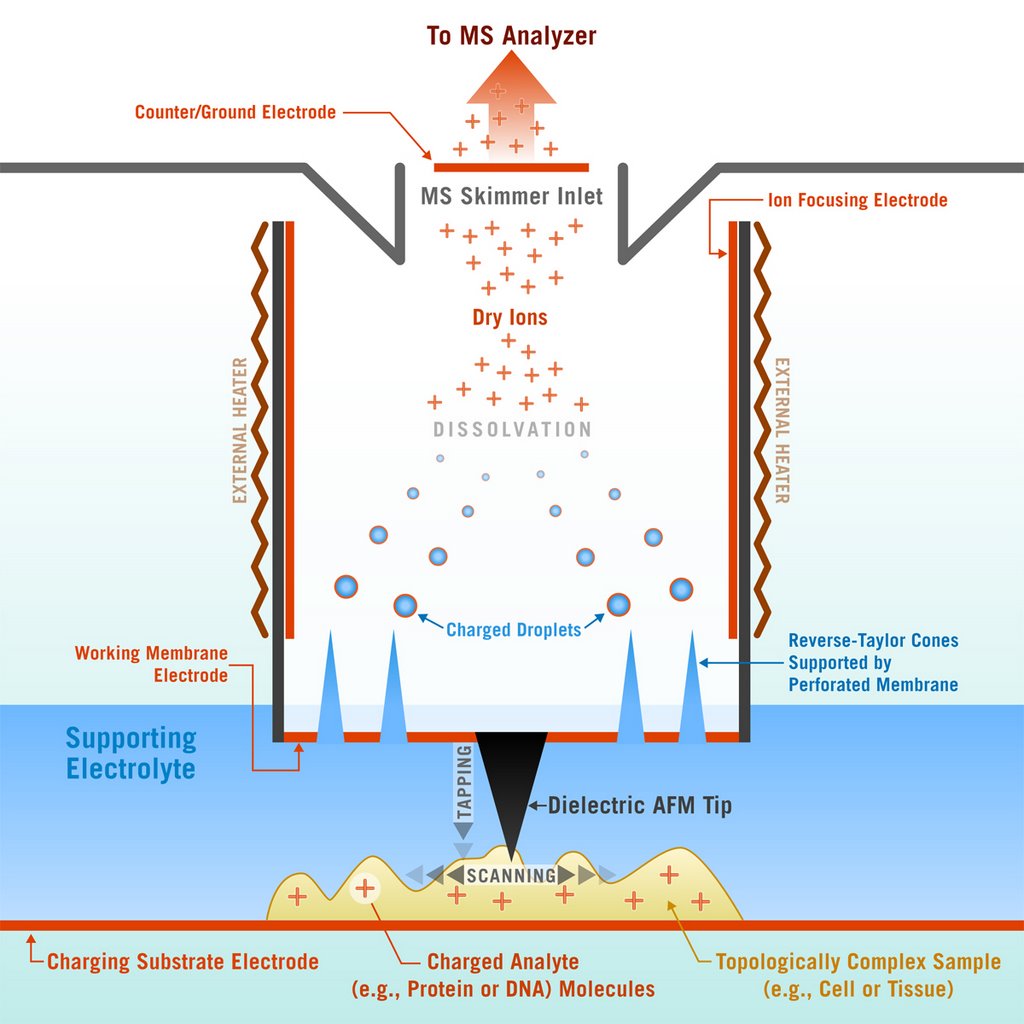
I think the way it works is the substrate gets a positive charge, the scanning tip has a negative charge. The scanning tip pulls molecules by charge, but also uses something called Taylor electrohydrodynamic focusing of jets to produce charged ions.
Wednesday, October 18, 2006
Why can't power plug configurations be universal?
We have all this excess noise in the lab, mostly from instrumentation, not from chatty n00bie employees. The lab I work in has two icp optical emission spectrometers and one icp mass spectrometer. Each instrument has a chiller and the mass spec also has a big ole vacuum pump. So, to prevent us from having to yell "WHAT" across the lab to one another, the boss had the idea to put the two chillers and the vacuum pump in another little room right next to the lab. That conversation was two months ago.
All we had to do was put some holes in the wall and get some longer power cords for everything. Sounds easy, but of course, it wasn't. The easy thing to do would be to plug the stuff in in the little room. But it all has to be on UPS and the only UPS outlets are in the lab. One chiller has a 110V power cord, not a problem at all, that is the normal plug in your house. The mass spec chiller has a 220V plug and it's from Europe. I never knew there were so many different power cord configurations. The power cord from the vacuum pump is also 220V, European, and different than the chiller cord from Europe.
There's a bunch of phone calls made, some red tape in the way, so we decide to move the mass spec right in front of the hole we made in the wall, so we don't need longer power cords. Genius.
Have a Guinness!
All we had to do was put some holes in the wall and get some longer power cords for everything. Sounds easy, but of course, it wasn't. The easy thing to do would be to plug the stuff in in the little room. But it all has to be on UPS and the only UPS outlets are in the lab. One chiller has a 110V power cord, not a problem at all, that is the normal plug in your house. The mass spec chiller has a 220V plug and it's from Europe. I never knew there were so many different power cord configurations. The power cord from the vacuum pump is also 220V, European, and different than the chiller cord from Europe.
There's a bunch of phone calls made, some red tape in the way, so we decide to move the mass spec right in front of the hole we made in the wall, so we don't need longer power cords. Genius.
Have a Guinness!
Saturday, September 16, 2006
Plumbing better than Mario and Luigi
Instead of dealing with large green pipes you have to jump into,
I have to deal with peristaltic pump tubing.

These little tubes are used to carry liquids into the mass spec. Other people use them too, such as chemists who use HPLC, IC, and SFA.
About twice a month, I have to run an analysis called FIAS or TTRA. Too make a long expanation short, the samples are mixed with buffer (pH 5.5) and internal standard (Y, In, Tb) all "online", then pushed through a column with an affinity resin that grabs the transition metals and lets the group I metals pass through. Then, the column is washed with acid, which rinses the transition metals into the mass spec which sorts them and counts them.

The way the literature describes to do this was unecessarily complicated. It explained using a sample loop and employing a water line to wash the column. It involved two valves, two pumps, four peristaltic pump lines, a double six-way valve, and finally the column.That's the way I did it and it worked for a while, but eventually it stopped working properly and I couldn't fix it. So instead of spending multiple days troubleshooting, I decided to just completely start over and re-think the whole thing.
I re-plumbed it much more simple and straight forward. Both valves, half of the double six-way valve, and the water line ended up being unnecessary. Now, loads of para-film were no longer needed to prevent leaks in the peristaltic pump tube connections. Also, less sample is used.



I have to deal with peristaltic pump tubing.

These little tubes are used to carry liquids into the mass spec. Other people use them too, such as chemists who use HPLC, IC, and SFA.
About twice a month, I have to run an analysis called FIAS or TTRA. Too make a long expanation short, the samples are mixed with buffer (pH 5.5) and internal standard (Y, In, Tb) all "online", then pushed through a column with an affinity resin that grabs the transition metals and lets the group I metals pass through. Then, the column is washed with acid, which rinses the transition metals into the mass spec which sorts them and counts them.

The way the literature describes to do this was unecessarily complicated. It explained using a sample loop and employing a water line to wash the column. It involved two valves, two pumps, four peristaltic pump lines, a double six-way valve, and finally the column.That's the way I did it and it worked for a while, but eventually it stopped working properly and I couldn't fix it. So instead of spending multiple days troubleshooting, I decided to just completely start over and re-think the whole thing.
I re-plumbed it much more simple and straight forward. Both valves, half of the double six-way valve, and the water line ended up being unnecessary. Now, loads of para-film were no longer needed to prevent leaks in the peristaltic pump tube connections. Also, less sample is used.


Wednesday, September 13, 2006
How to make a 1000 fold dilution
Making dilutions is an everyday thing when analyzing environmental samples. It's either diluting salt water samples by 10 because they have such a high ion content they suppress the plasma and cause internal standards to drop too low or diluting sediment samples because it takes more acid to digest them than is in the calibration standards.
Before entering into environmental analytical chemistry I would say to make a 1000 fold dilution, add 1000mL to 1 mL of what you're trying to dilute. But that's wrong, you add 999mL to 1mL of what you're trying to dilute. Reason is, it's already diluted once. If you start with pure copper for example, and stuck it in a beaker, then you would have to add 1000mL to dilute it 1000 fold. But if the copper is already dissolved in 1mL, then you would have to add 999mL to dilute to 1000 fold. I'm pretty sure you typically don't see pure metals, sitting around an environmental chemistry laboratory waiting to be dissolved into standards. It's much easier and cost effective to buy the metals already dissolved in water in nice, guaranteed concentrations, like 10,000ppm.
I can grow orchids --->


Before entering into environmental analytical chemistry I would say to make a 1000 fold dilution, add 1000mL to 1 mL of what you're trying to dilute. But that's wrong, you add 999mL to 1mL of what you're trying to dilute. Reason is, it's already diluted once. If you start with pure copper for example, and stuck it in a beaker, then you would have to add 1000mL to dilute it 1000 fold. But if the copper is already dissolved in 1mL, then you would have to add 999mL to dilute to 1000 fold. I'm pretty sure you typically don't see pure metals, sitting around an environmental chemistry laboratory waiting to be dissolved into standards. It's much easier and cost effective to buy the metals already dissolved in water in nice, guaranteed concentrations, like 10,000ppm.
I can grow orchids --->


Wednesday, August 30, 2006
How to get motivated for work
Someone asked me the other day how I got motivated for work. It didn't come out of the blue, we've been having some serious mass spec problems we just can't figure out. Turns out now, the technician can't really figure it out either. I replied I have to get paid, just kind of as a joke. I didn't really have time to think about it.
Maybe it's actually competition. Not just from people around me in the present, but people in the past. Stories of old chemists discovering things, having moments of clearity, and thinking "eureka!" actually motivates me. Maybe if I don't give in to working and feeling like a slave I'll get better at what I'm doing. The only way to solve a problem is to see the answer. It's hard to keep an open vision and look at things in a new way when you give into the stress of work problems.
So, to get motivated for work, seek inspiration from the past.
Maybe it's actually competition. Not just from people around me in the present, but people in the past. Stories of old chemists discovering things, having moments of clearity, and thinking "eureka!" actually motivates me. Maybe if I don't give in to working and feeling like a slave I'll get better at what I'm doing. The only way to solve a problem is to see the answer. It's hard to keep an open vision and look at things in a new way when you give into the stress of work problems.
So, to get motivated for work, seek inspiration from the past.
Tuesday, August 15, 2006
Unheard of ICPMS drift: Rabbit Pump Drift
Fellow mass spectrometerists, check this out. I'm running some calibration standards and all the sudden, from the 100ppb to the 200ppb, internal standard recovery changes from 100% to 80%. Naturally thought it was sample introduction; checked for clogs, changed cones, changed to new nebuliser, still same drift, sometimes after 5 cups. Then it eventually, 5 cups or so, it kind of creeps back up around 100%.
Today, I was looking at the real time display, in deep thought while I watched the 115In and 209Bi signal intertwine bouncing across the 20 in. flat screen when I figured it out. I accessed the accessory window and switched the peristaltic pump into rabbit mode[1]. Sure enough, the signal drops, probably around 80%, but just going off counts per second at the time. Let it equilibrate and switch it back to regular pump speed, 24%. Oh, man, took like 3 or 4 minutes for the counts to get back up to where they were before rabbit pump.
Edited ACL script, no rabbit pump, calibrated fine, loaded samples, got to name a new type of drift: Rabbit Pump Drift.
I also named Seesaw Drift, this is when the low mass internal standards drift up while the high mass internal standards drift down, or the other way around.
[1] This is what the software that runs the ICPMS calls max speed pumping. Good for washing the probe during rinse.
Today, I was looking at the real time display, in deep thought while I watched the 115In and 209Bi signal intertwine bouncing across the 20 in. flat screen when I figured it out. I accessed the accessory window and switched the peristaltic pump into rabbit mode[1]. Sure enough, the signal drops, probably around 80%, but just going off counts per second at the time. Let it equilibrate and switch it back to regular pump speed, 24%. Oh, man, took like 3 or 4 minutes for the counts to get back up to where they were before rabbit pump.
Edited ACL script, no rabbit pump, calibrated fine, loaded samples, got to name a new type of drift: Rabbit Pump Drift.
I also named Seesaw Drift, this is when the low mass internal standards drift up while the high mass internal standards drift down, or the other way around.
[1] This is what the software that runs the ICPMS calls max speed pumping. Good for washing the probe during rinse.
Monday, August 14, 2006
Typical drift problems
Links to help solve drift problems: link 1 , Perkin Elmer, Spectroscopy magazine, PlasmaChem.
It's not the sample introduction, it can't be.
 If you don't want to turn your head, translation for above, Wanted: Experienced Dog Catcher/Trapper to catch "one" small, smart, sneaky dog. Please call .... How great is that? Anybody up to it?
If you don't want to turn your head, translation for above, Wanted: Experienced Dog Catcher/Trapper to catch "one" small, smart, sneaky dog. Please call .... How great is that? Anybody up to it?

Random shot from the lab, do you think the guys who made this piece of equipment had a little laugh?
It's not the sample introduction, it can't be.
 If you don't want to turn your head, translation for above, Wanted: Experienced Dog Catcher/Trapper to catch "one" small, smart, sneaky dog. Please call .... How great is that? Anybody up to it?
If you don't want to turn your head, translation for above, Wanted: Experienced Dog Catcher/Trapper to catch "one" small, smart, sneaky dog. Please call .... How great is that? Anybody up to it?
Random shot from the lab, do you think the guys who made this piece of equipment had a little laugh?
Sunday, August 06, 2006
Cones before and after
A sample cone looks like this after you clean it:
 And a skimmer cone looks like this after some samples:
And a skimmer cone looks like this after some samples:
 I think that is permanganate built up on there. Anybody else have an idea?
I think that is permanganate built up on there. Anybody else have an idea?
 And a skimmer cone looks like this after some samples:
And a skimmer cone looks like this after some samples: I think that is permanganate built up on there. Anybody else have an idea?
I think that is permanganate built up on there. Anybody else have an idea?Friday, July 21, 2006
Slow down
It's hard to keep up in the blog world when your trying to move. I mean moving to a different residence. You always forget how much of a pain it is. I've got everything packed up in I-CHEM boxes swiped from work. Well they're not really swiped, they were getting thrown away.
Anyways, anyone know how to clean an extraction lens or re-install instrument controlling software without getting a headache?
Anyways, anyone know how to clean an extraction lens or re-install instrument controlling software without getting a headache?
Wednesday, July 05, 2006
Silver crashes
The other day I took on the rather long task of making a multi-element solution. A multi-element solution is exactly what it sounds like, a solution (a mixture of dissolved stuff), the dissolved stuff being many different elements. This particular solution was to be composed of 27 different metal elements such as chromium, arsenic, and manganese. One way to start making this solution is to dissolve different metallic salts, like As2O3, into a diluted acid (HNO3 or nitric acid) solution. But, I don't have to do that, we buy the metals already dissolved in acidic solutions.

So all I have to do is successfully pipette certain amounts like 10mL, 1mL, or 0.1mL. Then dilute the solution to 1 or 2L in a class A volumetric flask. It's not that hard if you got good lab technique, but you do have 27 chances to accidently add the wrong amount. Anyways, silver is always a little tricky to get into a solution. It always wants to precipitate out. I think it forms AgCl. The Cl anions come from the HCl I add to the solution along with the HNO3 to stabilize the metals. One way to prevent precipitation is to add the HCl last after the silver has already been added and is diluted throughout everything else. But in general, silver can't be put in a multi-element solution above 500ppb. Well for some reason I forgot everything I knew for 30 seconds and messed up the entire solution on the 27th metal, silver. As soon as I pipetted 0.5mL of a 1000ppm silver solution into about 1L of other dissolved metals + acid + water it crashed out into a white haze in the flask and eventually combined into what looks like normal table salt at the bottom of the volumetric flask, but I wouldn't put that salt on my fries.



So all I have to do is successfully pipette certain amounts like 10mL, 1mL, or 0.1mL. Then dilute the solution to 1 or 2L in a class A volumetric flask. It's not that hard if you got good lab technique, but you do have 27 chances to accidently add the wrong amount. Anyways, silver is always a little tricky to get into a solution. It always wants to precipitate out. I think it forms AgCl. The Cl anions come from the HCl I add to the solution along with the HNO3 to stabilize the metals. One way to prevent precipitation is to add the HCl last after the silver has already been added and is diluted throughout everything else. But in general, silver can't be put in a multi-element solution above 500ppb. Well for some reason I forgot everything I knew for 30 seconds and messed up the entire solution on the 27th metal, silver. As soon as I pipetted 0.5mL of a 1000ppm silver solution into about 1L of other dissolved metals + acid + water it crashed out into a white haze in the flask and eventually combined into what looks like normal table salt at the bottom of the volumetric flask, but I wouldn't put that salt on my fries.


Sunday, June 25, 2006
Plasma in the window
You know a lot of people play Sudoku and work crossword puzzles to exercise their mind. Any chemists out there ever try to draw out syntheses for natural products or any other molecules to gain extra brain power? Here's a shot of the plasma from the lab.

The color of the plamsa is actually white, but the window is tinted green with something that prevents the UV light from harming our eyes. The ultraviolet light is emitted from the metals in the water samples after they gain energy from the plasma. There is actually no reason for the window at all, it just looks cool.

The color of the plamsa is actually white, but the window is tinted green with something that prevents the UV light from harming our eyes. The ultraviolet light is emitted from the metals in the water samples after they gain energy from the plasma. There is actually no reason for the window at all, it just looks cool.
Monday, June 19, 2006
Quality Control Explained
Quality control in ensuring your product, or scientific result falls in between a set of guidelines determined by the necessary amount of accuracy and precision required. It allows a customer to know how reliable a scientific result is.
For instance, say you want to analyze some water samples from off the coast of Louisiana to check if there is any lead contamination because the battery factory was flooded and damaged from a hurricane. To start with, you might want to take samples in various locations and record some type of data like distance from shore and maybe depth of sample taken along with the pH of the water. To tell that the guy or girl sampling the water hasn't contaminated it themselves, they'll need to take some DI water up with their water sampling equipment and bottle that up to go with all the sea water samples.
Then, it'll get back to the lab and it will have to be prepared to be analyzed, the preparer will include another blank with the batch of sea water samples to ensure that he or his lab did not contaminate the samples. These blank samples are representative of the whole batch of samples, if they have any lead in them, then all the other sea water samples would be expected to have the same amount. That's why you gotta keep it all clean, with a mass spec you're usually looking for pretty low amounts.
To be continued... as a series or possibly just a trilogy.
For instance, say you want to analyze some water samples from off the coast of Louisiana to check if there is any lead contamination because the battery factory was flooded and damaged from a hurricane. To start with, you might want to take samples in various locations and record some type of data like distance from shore and maybe depth of sample taken along with the pH of the water. To tell that the guy or girl sampling the water hasn't contaminated it themselves, they'll need to take some DI water up with their water sampling equipment and bottle that up to go with all the sea water samples.
Then, it'll get back to the lab and it will have to be prepared to be analyzed, the preparer will include another blank with the batch of sea water samples to ensure that he or his lab did not contaminate the samples. These blank samples are representative of the whole batch of samples, if they have any lead in them, then all the other sea water samples would be expected to have the same amount. That's why you gotta keep it all clean, with a mass spec you're usually looking for pretty low amounts.
To be continued... as a series or possibly just a trilogy.
Wednesday, June 14, 2006
Alpha hydroxy garbage
This is kinda funny. After studying organic chemistry, whenever those commercials came on TV about "alpha hydroxys" in skin creams and stuff I would laugh and say that doesn't mean anything. I tried to explain to my girlfriend that the term alpha hydroxy just describes a location on a molecule, like the next carbon over from something has an alcohol bonded to it. In other words, the alcohol (hydroxy) is alpha (next to) something, but you can't really tell what that something is when you say alpha hydroxy.
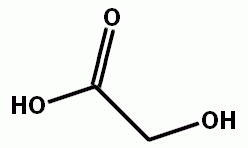
Glycolic acid
Some of the commercials say "alpha hydroxy acids". This makes much more sense, turnes out, there is a group of molecules called alpha hydroxy acids. And they do have an effect on skin.
Another thing that tickles my fancy is seeing all those little multi-colored beads mixed in and suspended in the face wash or shampoo. That is actually some hot research because just the way it looks sitting on the grocery store shelf makes people buy it. So there were actually scientist out there trying to find out how to suspend little colored balls in a hair gel, making and spending loads of money. It's just the way it goes. Kinda seems like a waste of scientific talent to me, maybe the project only took a couple years.
Next up, beta hydroxy acids.
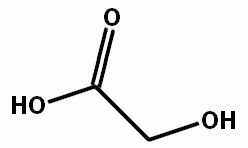
Glycolic acid
Some of the commercials say "alpha hydroxy acids". This makes much more sense, turnes out, there is a group of molecules called alpha hydroxy acids. And they do have an effect on skin.
Another thing that tickles my fancy is seeing all those little multi-colored beads mixed in and suspended in the face wash or shampoo. That is actually some hot research because just the way it looks sitting on the grocery store shelf makes people buy it. So there were actually scientist out there trying to find out how to suspend little colored balls in a hair gel, making and spending loads of money. It's just the way it goes. Kinda seems like a waste of scientific talent to me, maybe the project only took a couple years.
Next up, beta hydroxy acids.
Friday, June 09, 2006
New calibration standards
Calibration standards have to be made in order for the mass spec to correlate the intensity of the ion signal to concentration. You can't just turn it on and stick a sample in and get a result of 50ppb Cd like on CSI. (A mass spec finds atoms of interest in big piles of random atoms). What would happen if you did that is you would get some random result such as 564,000 CPS (counts per second). When I "calibrate" the instrument by analyzing a 25ppb (10,000CPS), 50ppb (20,000CPS), and 100ppb (40,000CPS) at the beggining of the run, then the instrument knows what 564,000 CPS is. Yes, if you graphed intensity (CPS) vs. concentration (ppb), it would be linear, that's how it works. Technically, 564,000 CPS is obviously overcal in this example and would have to be rerun diluted.

Back to the calibration standards, or multi-element solutions, these are a pain to make. You take bottles off the shelf such as 10,000ppm Sb, it's dissolved in acidified D.I. water from some kind of compound like Sb2O3. You take about 20 to 30 of these bottles off the shelf including things like Tl, Pb, Mn, and Ag. The tricky part is dispensing a very small and precise amount of each element into a 250 or 500mL volumetric flask to arrive at a solution where each element is exactly 50ppb. There is a quite simple calculation to find out how much 10,000ppm Sb to add to a 500mL flask to get 50 ppb. M1*V1 = M2*V2
(10,000ppm Sb)*(x mL) = (0.050ppm)*(500mL)
x = 0.0025mL or 2.5uL

In this case I would have to make an intermediate of say 10ppm and then dilute that down to 50ppb because our pipettes don't go down to 2.5uL.

Then I have your basic quality control guidelines and if the solution I made doesn't "pass", it has to be remade. One can see where it can become frustrating. And besides just calibration standards, I need to make a second source calibration check, an interference check solution, as well as all the spiking solutions used in the lab.
Thursday, June 08, 2006
On global warming from a sceptical chemist
I got into a very interesting and controversial conversation while on a trip to Chicago with a jaunt to Green Bay last week.

The topic of global warming came up from someone who believes it is happening and that something must be done about it. I made a little remark about how I was still somewhat sceptical of the whole global warming argument. And the reaction was surprisal, especially me being a scientist and everything. They went on to talk about how scientist views are being put down or hushed by Washington government. That in itself is very alarming and really requires its own post.
It gets real messy when politics get mixed in with science. You really don't know whose views are swayed by whatever. Really, what I think the problem is, I can't find information on the subject that I don't feel has been politically influenced. What I have read is mostly from science articles on the web that are conflicting. That is how it appears to me, is that scientist actually don't agree on global warming.
Instead of commenting about how I know nothing about global warming (I admit, I'm not an expert), please give me a link to some scientific literature about global warming that you believe to be the truth. I mean the facts, some evidence, you know, experimentation. I'll take a sceptical look at computer models.
By the way, I noticed I made an appearance on the Mass Spectrometry Blog.
An interesting blog by a fellow mass spectrometerist.
Cutting edge literature_, Chemistry facts_, Rants and raves_,

The topic of global warming came up from someone who believes it is happening and that something must be done about it. I made a little remark about how I was still somewhat sceptical of the whole global warming argument. And the reaction was surprisal, especially me being a scientist and everything. They went on to talk about how scientist views are being put down or hushed by Washington government. That in itself is very alarming and really requires its own post.
It gets real messy when politics get mixed in with science. You really don't know whose views are swayed by whatever. Really, what I think the problem is, I can't find information on the subject that I don't feel has been politically influenced. What I have read is mostly from science articles on the web that are conflicting. That is how it appears to me, is that scientist actually don't agree on global warming.
Instead of commenting about how I know nothing about global warming (I admit, I'm not an expert), please give me a link to some scientific literature about global warming that you believe to be the truth. I mean the facts, some evidence, you know, experimentation. I'll take a sceptical look at computer models.
By the way, I noticed I made an appearance on the Mass Spectrometry Blog.
An interesting blog by a fellow mass spectrometerist.
Cutting edge literature_, Chemistry facts_, Rants and raves_,
Tuesday, May 30, 2006
Everybody should read this

I read a story on Wired news about garage chemistry. I wish they still sold chemistry sets that I thought would be fun to use. Like all the chemicals and equipment necessary to isolate and characterize natural products. I think I would specialize in marine natural products, they don't seem to be a passing fad.
Organic chemistry_, Cutting edge literature_
Thursday, May 25, 2006
The Golden Run
Last night's run was what we analytical chemists call a golden run. This means that all the analytes that I needed to analyze for, for each sample, had the yellow highlighter run over them on the cover page of the lab report. The yellow highlighter means that the result is good to go and I don't need to reanalyze. This has only happened one other time in the year I have been running the mass spec.
Talk about staying steady, the mass spec was on point for at least 12 to 14 hours. Now, sales people for mass specs say their instruments can stay "in cal." for like 2 or 3 days. What they don't tell is they are analyzing nice clean pretty sample in D.I. water. Get some real samples in there with loads of salt or suspended solids and your instrument might not make it past the first sample like that.
In the lab_,
Talk about staying steady, the mass spec was on point for at least 12 to 14 hours. Now, sales people for mass specs say their instruments can stay "in cal." for like 2 or 3 days. What they don't tell is they are analyzing nice clean pretty sample in D.I. water. Get some real samples in there with loads of salt or suspended solids and your instrument might not make it past the first sample like that.
In the lab_,
Tuesday, May 23, 2006
Backwards chromatography with Mass Spec
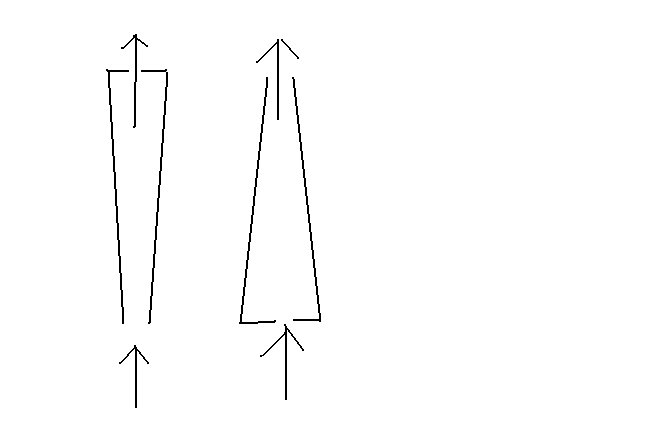
I've been trying to run the FIAS analysis for three days now, the column just doesn't seem to be doing its job. I think the problem might be that I have it backwards. I believe the way the flow should be is into the large end and out the small end. This way, the front of the column will bind the most metals, then, when the flow reverses to rinse the metals off the column with dilute acid, the metals will be at the end of the column, so they come off together.
Subscribe to:
Posts (Atom)




